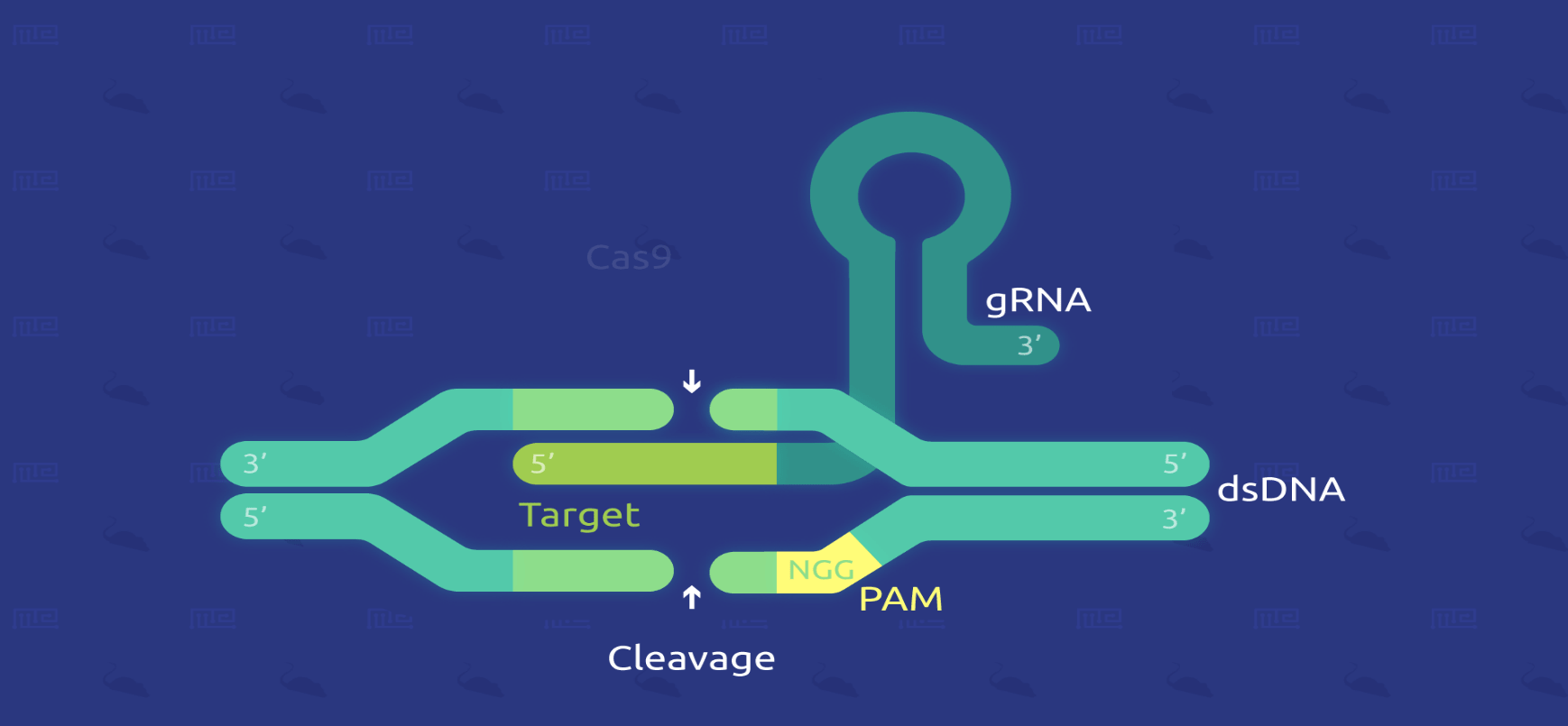
روشهای دستکاری هدفمند ژنوم
15 خرداد 1399
کلونینگ مولکولی (Molecular Cloning)
کلونینگ مولکولی به فرآیندی اطلاق میشود که توسط آن مولکولهای نوترکیب DNA تولید، به یک ارگانیسم میزبان انتقال داده شده، و در آنجا تکثیر مییابند. یک واکنش کلونینگ مولکولی معمولاً شامل بر دو جزء اصلی زیر میباشد:
۱- قطعه DNA مورد نظر یا اینسرت ( Insertیا DNA fragment of interest)؛
۲- وکتور/ پلاسمید که شامل تمام اجزاء لازم برای تکثیر در ارگانیسم میزبان است.
DNA مورد نظر یا ژن هدف
DNA مورد نظر (یا ژن هدف؛ Gene of interest) میتواند یک ژن، عناصر تنظیمکننده یا اپرون، یک پروموتر یا هر قطعه دیگری باشد. آن را میتوان با روشهای مختلفی برای کلون کردن آماده کرد:
- DNA مورد نظر را میتوان با استفاده از آنزیمهای محدودکننده از یک منبعِ DNAی دیگر مانند وکتور/ پلاسمید یا DNA ژنومی برش زد.
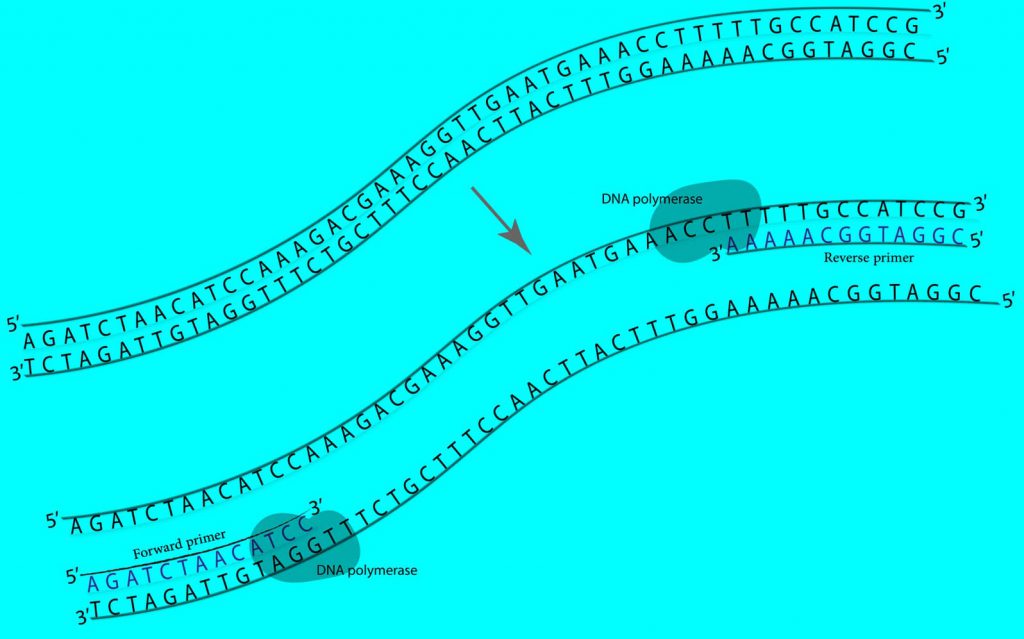
- با استفاده از واکنش زنجیرهای پلیمراز (PCR)، قطعه DNAی مورد نظر را میتوان تکثیر کرد.
- قطعه DNAی مورد نظر را میتوان با به هم چسباندن (Annealing) اولیگونوکلئوتیدهای تک رشتهای مونتاژ کرد.
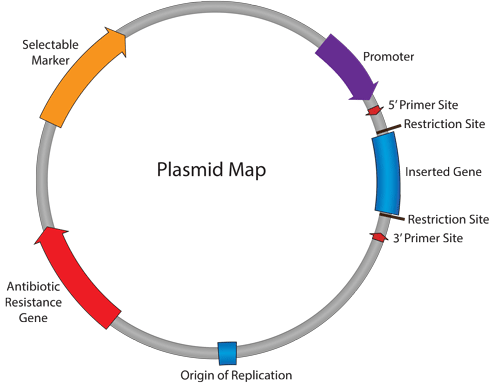
وکتور/ پلاسمید
پلاسمید (پلازمید) یک قطعه کوچک و حلقوی از DNA است که در داخل سلول میزبان (مجزا از DNA کروموزومی یا ژنومی) تکثیر مییابد. اغلب پلاسمیدهای رایج مورد استفاده در تکنولوژیِ DNA نوترکیب، به منظور مطالعه و دستکاری ژنها بهینه شدهاند. به عنوان مثال بیشتر پلاسمیدها در باکتری E. coli تکثیر میشوند و دارای اندازهی نسبتاً کوچک (∼ 3000 تا 6000 جفت باز) برای دستکاری آسان میباشند. با اتصال فیزیکیِ DNA مورد نظر به وکتور/ پلاسمید، از طریق پیوندهای فسفودی استر، DNA مورد نظر بخشی از پلاسمیدِ نوترکیب جدید میشود.
وکتور/ پلاسمیدها این امکان را فراهم میکنند تا DNA مورد نظر در مقادیر زیادی (در سلول میزبان) کپی شوند، و اغلب عناصر کنترلی لازم برای هدایت رونویسی و ترجمه DNA کلون شده را فراهم میکنند. به این ترتیب وکتورها برای بسیاری از روشهای مولکولی مانند بیان پروتئین، بررسی بیان ژن و آنالیز عملکردی مولکولهای زیستی، ابزارهای مناسب و کاربردی هستند. به طور معمول پلاسمیدها حاوی حداقل توالی DNA لازم برای این منظور هستند؛ شامل منشاء همانندسازی DNA (DNA replication origin)، ژن مقاومت به آنتیبیوتیک (Antibiotic-resistance gene) و ناحیهای که در آن، قطعات DNA خارجی را میتوان وارد کرد.
هنگامی که یک پلاسمید در E. coli بصورت خارج کروموزومی (Extrachromosomal) وجود دارد، به طور مستقل تکثیر میشود و به سلولهای دختر حاصل از تقسیم، به ارث میرسد. این سلولهای دختری، حاوی اطلاعات ژنتیکی مشابه با سلول مادری خود هستند و بنابراین به آنها کلون سلول اصلی (Clones of the original cell) گفته میشود. DNA پلاسمیدی به طور مشابه، DNA کلون شده (Cloned DNA) نامیده میشود. فرایندِ تولید چندین نسخه یکسان از یک مولکول DNAی نوترکیب نیز، به عنوان کلونینگ (کلونسازی)DNA یا کلونینگ مولکولی (DNA or molecular cloning) شناخته شده است.
فرآیند کلونینگ مولکولی به دانشمندان این امکان را میدهد تا کروموزومها و ژنها را مورد مطالعه قرار دهند. امروزه دانشمندان میتوانند ژنها و سایر عناصر ژنتیکی را به راحتی با استفاده از پلاسمیدهایی که بطور خاص مهندسی شدهاند، مورد مطالعه و دستکاری قرار دهند و به این ترتیب پلاسمیدها تبدیل به ابزارهای قدرتمندی در ژنتیک مولکولی شدهاند.
در جدول زیر تعدادی از عناصر اصلی به کار رفته در پلاسمیدها توضیح داده شدهاند:
| عناصر اصلی پلاسمید | توضیح |
|---|---|
| منشا همانندسازی Origin of Replication (ori) | توالی DNAای که با فراخواندن ماشین همانندسازی DNA، تکثیر پلاسمید (توسط سلول میزبان) را هدایت میکند. |
| ژن مقاومت به آنتیبیوتیک Antibiotic Resistance Gene | امکان انتخاب سلولهای حاوی پلاسمید را (با استفاده از مزیت بقا برای میزبان) فراهم میکند. در مورد استفاده در باکتری،: به دلیل بار اضافهای که برای تکثیر پلاسمید (علاوه بر DNAی ژنومی میزبان) در هر بار تقسیم در باکتریهای حاوی پلاسمید وجود دارد، این باکتریها، نسبت به باکتریهایی که پلاسیمد را دریافت نکردهاند دارای سرعت تقسیم کمتری هستند (یعنی برای کپی کردن این DNA اضافی زمان بیشتری طول میکشد). بنابراین برای اطمینان از حفظ DNA پلاسمیدی در جمعیت باکتریها، یک ژن مقاومت به آنتی بیوتیک در پلاسمید وجود دارد و باکتریهای حاوی این پلاسمید، در حضور آنتیبیوتیک میتوانند رشد کنند و آنهایی که فاقد پلاسمید هستند دوام نمی آورند. ژن مقاومت آنتی بیوتیکی باید تحت کنترل یک پروموتر باکتریایی باشد تا با استفاده از دستگاه رونویسی باکتریایی در میزبان باکتری بیان شود. |
| جایگاه برش چندگانه Multiple Cloning Site (MCS) | قطعهی كوتاهی از DNA كه حاوی چندین سایت شناسایی برای برش آنزیمهای محدودكننده است و وارد کردن DNAی موردنظر با برش آنزیمی و سپس اتصال (Ligaton) را تسهیل میکند. در پلاسمیدهای بیانی (Expression plasmids)، MCS اغلب در پایین دستِ پروموتر قرار دارد، به گونهای که وقتی یک ژن در MCS وارد گردد، بیان آن توسط پروموتر هدایت میشود. این سایتهای شناسایی آنزیمهای محدود کننده در MCS، منحصر به فرد هستند و در جای دیگری در بدنه پلاسمید قرار ندارند. برای اطلاعات بیشتر در مورد آنزیمهای محدود کننده، به مقالات دیگر در همین وب سایت مراجعه کنید. |
| DNAی مورد نظر (Insert) | پروموتر یا قطعه DNAی دیگری است که در MCS کلون میگردد و معمولاً یک عنصر ژنتیکی است (که با استفاده از پلاسمید) هدف مطالعه است. |
| ناحیه پروموتری Promoter Region | رونویسی از DNAی مورد نظر را هدایت میکند. پروموتر به منظور فراخواندن دستگاه رونویسیِ ارگانیسم یا گروهی از موجودات خاص طراحی شده است. به این معنی که اگر پلاسمید در نظر گرفته شده برای استفاده در سلولهای انسانی باشد، پروموتر باید یک توالی خاص تنظیمی در انسان یا پستانداران باشد. پروموتر همچنین میتواند بیان مختص سلول (Cell-specific expression) خاصی را هدایت کند، که در این صورت از یک پروموتر مختص بافت (Tissue-specific promoter)، به عنوان مثال از یک پروموتر مختص کبد استفاده میشود. قدرت پروموتر برای کنترل سطح بیان Insert نیز مهم است (یعنی یک پروموتر قوی بیان بالا، در حالی که پروموتر ضعیفتر میتواند سطح بیان پایین را هدایت کند). |
| نشانگر انتخابی Selectable Marker | نشانگر انتخابی برای انتخاب سلولهای میزبان است که پلاسمید را به منظور بیان Insert، با موفقیت دریافت کردهاند. این انتخاب، متفاوت از انتخاب سلولهای باکتریایی است که پلاسمید با هدف همانندسازی، دریافت کردهاند. نشانگر انتخابی امکان انتخاب جمعیت سلولهایی که پلاسمید را گرفتهاند و میتوانند برای مطالعه Insert مورد استفاده قرار گیرند، را فراهم میکند. مارکر انتخابی معمولا ژن مقاومت آنتی بیوتیک (تحت کنترل یک پروموتر غیر باکتریایی) و یا یک پروتئین فلورسنت است. |
| محل اتصال پرایمر Primer Binding Site | پرایمر یک توالی کوتاه تک رشتهای DNA است که به عنوان نقطه شروع برای تکثیر با PCR یا تعیین توالیِ DNA (DNA sequencing) پلاسمید استفاده میشود. از پرایمرها برای تأیید توالی Insert یا مناطق دیگر پلاسمید میتوان استفاده کرد. پرایمرهای رایج را میتوانید در لیست پرایمرهای تعیین توالیِ در سایت دنازیست آسیا بررسی کنید. |
برای خطی کردن وکتور/ پلاسمید مورد نظر بمنظور آمادهسازی برای کلون کردن اینسرت، از آنزیمهای محدودکننده یا PCR استفاده میشود.
در طی فرآیند کلونینگ، انتهای DNA مورد نظر و وکتور/ پلاسمید باید اصلاح گردد تا برای اتصال توسط DNA لیگاز (DNA ligase)، ریکامبیناز (Recombinase)، توپوایزومراز (Topoisomerase) یا مکانیسمهای داخل سلولیِ ترمیم DNA، هر دو انتهای آنها باهم سازگار شوند. برای این منظور معمولا از آنزیمهایی مانند نوکلئازها، فسفاتازها، کینازها و یا لیگازها استفاده میکنند.
امروزه روشهای زیادی برای سادهتر کردن و استانداردسازی انجام کلونینگ مولکولی معرفی شدهاند و همچنین کیتهای جدیدی نیز برای این منظور توسط شرکتهای مختلف ساخته شدهاند. این موضوع در یادداشتهای بعدی توضیح داده خواهد شد. در این یادداشت تنها روش مرسوم و کلاسیک کلونینگ مولکولی توضیح داده میشود.
کلونینگ مرسوم یا کلاسیک (Traditional Cloning)
اگرچه امروزه نسلهای جدیدی از مونتاژ DNA (DNA assembly) و کلونینگ مولکولی معرفی شدهاند، با این حال استفاده از روشهای کلاسیک کلونینگ (Traditional Cloning) هنوز در بسیاری آزمایشگاهها متداول است.
در کلونینگ کلاسیک از آنزیمهای محدودکننده برای خطی (Linearize) کردن وکتور/ پلاسمید و خارج کردن قطعه DNA مورد نظر از یک منبع DNA استفاده میشود، بطوری که ضمن برش با این آنزیمها، انتهاهای سازگار ایجاد میگردد. پس از تخلیص وکتور خطی و DNA مورد نظر (Insert)، با فعالیت آنزیمی DNA-لیگاز (DNA ligase)، این دو بهم متصل شده و وکتور نوترکیبِ جدید بمنظور تکثیر مولکول نوترکیب، به یک میزبان E. coli وارد (Transform) میشود.
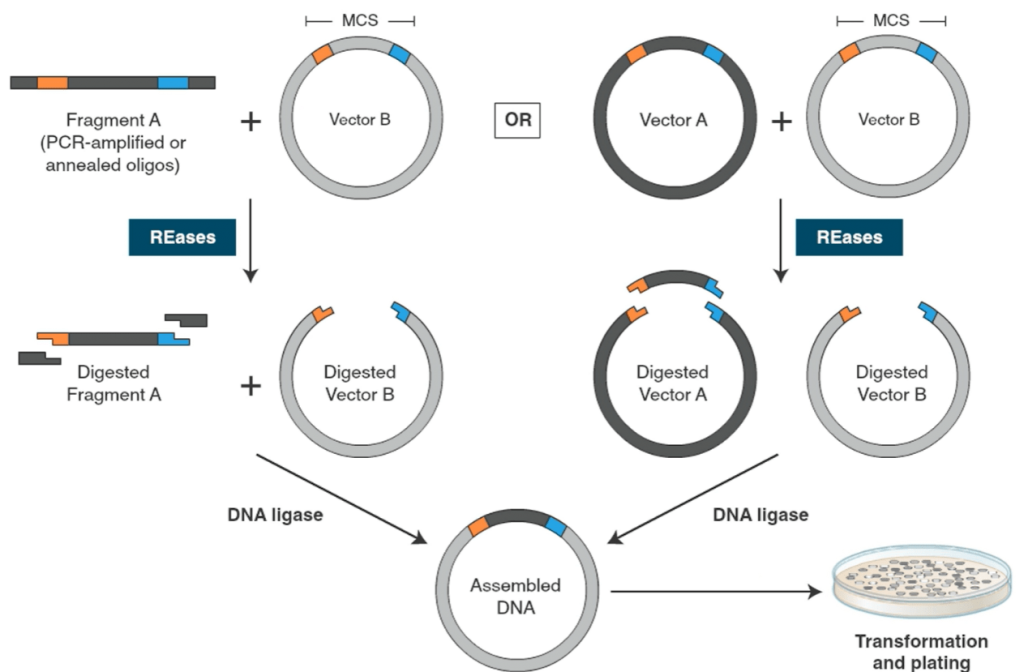
صرف نظر از این که کدام روش کلونینگ انتخاب شده باشد، با پیروی از پروتکلهای خوب و مناسب در آزمایشگاه میتوان این فرایند را مؤثرتر و موفقتر انجام داد. در ادامه به معرفی نکات مهم در یک پروژه کلونینگ و همچنین مراحل انجام کلونینگ کلاسیک میپردازیم.
۸ نکته مهم برای داشتن یک کلونینگ مولکولی موفق
۱- زمان کافی برای طراحی آزمایشهای خود اختصاص دهید
توجه به جزئیات هنگام طراحی و برنامهریزی برای یک پروژه کلونینگ ضروری است. با درک کاملی از روشهای مورد استفاده و توالیهای تولید شده، اطمینان حاصل کنید که طراحی شما صحیح و دقیق است. به اتصال توالیها و اثر آن بر روی چارچوب خوانش (Open reading frame; ORF) هر توالیِ کدکننده توجه کنید. به جایگاههای شناسایی آنزیمهای محدودکننده (Restriction site of restriction enzyme)، هم در وکتور و هم در DNA مورد نظر (Insert)، قبل از طراحی پرایمرهای PCR (که حاوی سایتهای شناسایی مشابه با آنچه در کلونینگ استفاده میشوند، هستند) دقت کنید. مطمئن شوید که نشانگر انتخابی آنتیبیوتیکی (Antibiotic selective marker ) در وکتور، با نژاد میزبان انتخابی سازگار باشد. پیشنهاد میشود قبل از شروع کار در آزمایشگاه، مراحل انجام پروژه کلونینگ خود را در نرم افزارهای آنلاین و یا آفلاین مخصوص این کار، شبیهسازی کنید.
۲- با یک DNA تمیز و در غلظت مناسب شروع کنید
اطمینان از اینکه منبع DNA شما عاری از آلایندهها، از جمله نوکلئازها و فعالیتهای آنزیمی ناخواسته است، مهم میباشد. استفاده از کیتهای مبتنی بر ستون مانند کیتهای استخراج از محصول PCR، و استخراج از ژل ستونی برای خالصسازی DNA، روش خوب و مناسبی است. قبل از دستکاری DNA، تمام حلالها از جمله فنل، کلروفرم و اتانول را کاملاً حذف کنید. اطمینان حاصل کنید که شستشوی نهایی و خروج DNA از ستونها، با بافر عاری از نمک انجام شده باشد. به این ترتیب مانع از مهار مراحل پایین دست مانند برش با آنزیمهای محدودکننده و یا تکثیر با PCR میشوید. برای تکنیک مورد استفاده در پروژه کلونینگ خود مقدار کافی از DNA وارد واکنش کنید. بمنظور برش با آنزیمهای محدودکننده، اغلب بین 2/0 تا 2 مایکروگرم نیاز است، در حالیکه تنها مقادیر بسیار کمی (در حد نانوگرم) از DNAی الگو برای انجام PCR کافی است.
کیت استخراج پلاسمید (روش ستونی)
۱,۵۱۷,۰۰۰ تومان–۲,۷۰۷,۰۰۰ تومان
۳- برش با آنزیمهای محدودکننده را با دقت انجام دهید
مهم است که هنگام برش DNA، واکنش هضم با آنزیم را به درستی تنظیم کنید. حجم واکنش باید با مرحله پایین دست، سازگار باشد به عنوان مثال باید کمتر از حجم چاهک ژل آگارز ِمورد استفاده برای جدا کردن قطعات DNA باشد. اغلب حجم یک واکنش کلونینگ کلاسیک، بین 20 تا 50 مایکرولیتر است. حجم آنزیم (های) محدودکننده اضافه شده نباید بیش از 10٪ از حجم کل واکنش باشد، تا اطمینان حاصل شود که غلظت گلیسرول زیر 5٪ در واکنش باقی میماند. این نکته برای به حداقل رساندن فعالیت ناخواسته و غیر اختصاصی آنزیمهای محدود کننده (Star activity) مهم و ضروری است.
۴- به انتهاهای ایجاد شده توجه کنید
با فرض اینکه انتهاهای ایجاد شده برای اتصال سازگار باشند، یعنی دارای انتهای چسبندهی (Overhang یا Sticky end) مکمل یا انتهای صاف (Blunt end) باشند، DNAی مورد نظر و وکتور بدون اصلاحِ بیشتر برای کلونینگ آماده هستند. اما اگر انتهاهای آنها سازگار نباشند، باید با استفاده از روشهای مناسب اصلاح شوند (به عنوان مثال از blunting reagents، فسفاتازها و غیره استفاده شود).
اگر برای آمادهسازی انتهاهای DNA، از PCR و DNAپلیمرازِ Taq (Taq DNA Polymerase) استفاده میشود، یک نوکلئوتید آدنین (A) اضافی در انتهای 3 پرایم آن باقی میماند. استفاده از DNAپلیمرازهای High-fidelity (High-fidelity DNA polymerases)، انتهای صاف ایجاد میکند. انجام PCR، با استفاده از پرایمرهای تجاری استاندارد، قطعات غیرفسفریله تولید میکند، مگر اینکه پرایمرها در انتهای 5 پرایم خود فسفریله باشند. محصول PCR ممکن است قبل از اتصال به یک وکتور دفسفریله، نیاز داشته باشد تا با یک آنزیم کیناز (مانند T4 Polynucleotide Kinase; T4 PNK) تیمار شود تا یک گروه فسفات در انتهای 5 پرایم آن اضافه گردد.
۵- قبل از اتصال وکتور به DNA مورد نظر، هر دو جزء را تمیز کنید
استفاده از کیتهای استخراج محصول PCR (یا PCR & DNA Cleanup Kit)، در مواردی که قطعه DNA مورد نظر توسط PCR تکثیر مییابد، میتواند نتایج کلونینگ شما را به طرز چشمگیری بهبود بخشد. همچنین استفاده از الکتروفورز ژل آگارز برای جداسازیِ قطعه DNA مورد نظر از سایر قطعات ناخواسته (پس از هضم با آنزیم) متداول است. در این روش قطعه DNA مورد نظر در زیر نور UV از روی ژل برش زده میشود و در نهایت با استفاده از کیت استخراج ژل قابل بازیابی است. استفاده از طول موج بلند (365 نانومتر) برای به حداقل رساندن هرگونه آسیب DNA ناشی از اشعه ماوراء بنفش پیشنهاد میگردد.
۶- مقادیر قطعات (DNA) خود را اندازه گیری کنید
با روشهای ساده سنجش کمی مانند الکتروفورز ژل با استانداردهای مشخص (Gel electrophoresis with mass standards) یا طیف سنجی با استفاده از اسپکتروفتومترهای با ورودی کم (مانند نانودراپ)، از مناسب بودن مقادیر مواد برای انجام واکنش اتصال (Ligation reaction) اطمینان حاصل کنید.
۷- دستورالعملهای شرکتهای سازنده را برای واکنش اتصال (joining/ligation) دنبال کنید
برای کلونینگ معمولی دستورالعملهای تعیینشده توسط شرکت تهیهکننده لیگاز را دنبال کنید. اگر نسبت مولی 1 به 3 قطعه مورد نظر (Insert) به وکتور توصیه میشود، در ابتدا برای رسیدن به بهترین نتیجه این نسبت را امتحان کنید. استفاده از این نسبت 1 به 3 ثابت نیست و بسته به پیچیدگی پروژه کلونینگ شما میتواند تغییر کند.
سایتهای مختلفی برای محاسبهی نسبت مولیِ قطعه مورد نظر (Insert) به وکتور وجود دارد، که برای نمونه لینک تعدادی از آنها در زیر آورده شده است.
https://nebiocalculator.neb.com/#!/ligation
http://www.insilico.uni-duesseldorf.de/Lig_Input.html
https://worldwide.promega.com/resources/tools/biomath
۸- از سلولهای مستعد (Competent cell) متناسب با نیازهای خود استفاده کنید
اگرچه پروتکلهای زیادی برای مستعدسازی سلول میزبان وجود دارد، ولی استفاده از سلولهای مستعد تجاری میتواند باعث صرفهجویی در وقت و منابع شما شود و کلونینگ شما قابل تکرار خواهد بود.
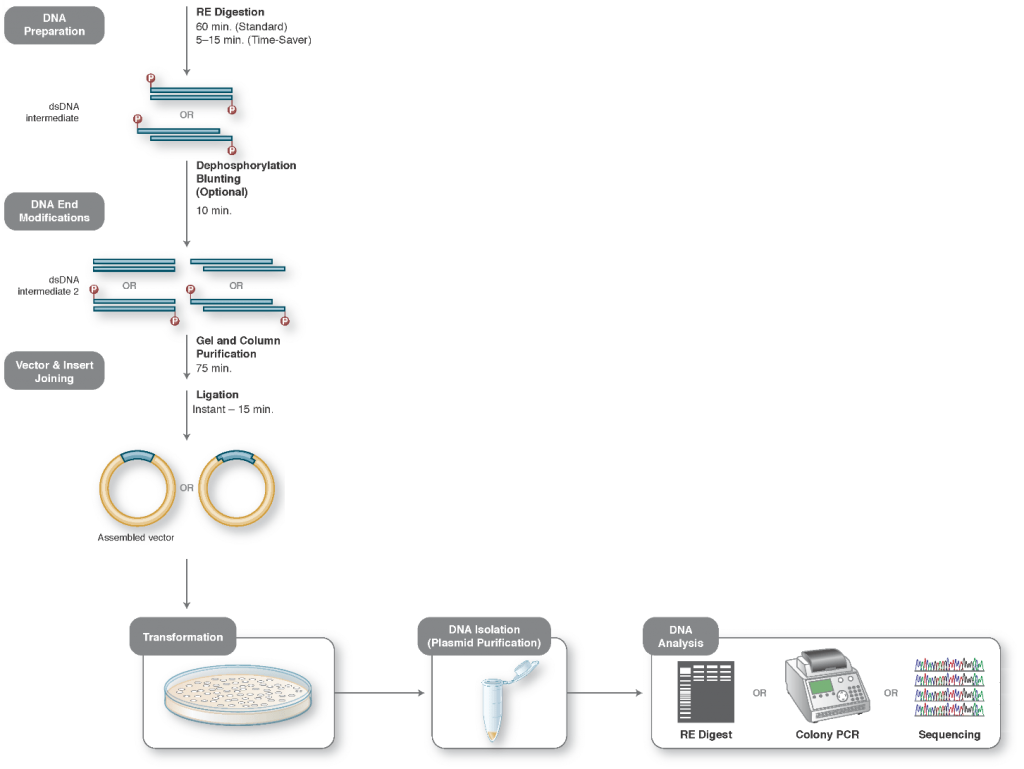
مراحل کار کلونینگ مولکولی کلاسیک

۱- برش DNA
بمنظور برش پلاسمید پذیرنده (Recipient plasmid) و پلاسمید دهنده (Donor plasmid)، آنزیمهای محدودکنندهی مناسبی را انتخاب کنید. از آنجا که مقداری DNA در مرحلهی استخراج ژل از دست میرود، بنابراین هضم آنزیمی مقادیر کافی از DNA اهیمت زیادی دارد. 5/1 تا 2 مایکروگرم از پلاسمید دهنده و 1 مایکروگرم از پلاسمید گیرنده توصیه میشود. همچنین برش کامل پلاسمید گیرنده مهم است. بنابراین مدت زمان هضم آنزیمی بایستی حداقل 4 ساعت و حتی تا یک شب کامل (overnight) باشد.
اگر قصد دارید فقط از یک آنزیم محدودکننده یا آنزیمهایی استفاده کنید که انتهاهای صاف و یا چسبندهی مکمل پس از هضم ایجاد میکنند، برای جلوگیری از حلقوی شدن مجدد (یا Self-ligation) پلاسمید گیرنده، باید از فسفاتاز استفاده کنید. برای این منظور پلاسمید گیرنده برش خورده قبل از مرحلهی اتصال (Ligation) و یا قبل از استخراج ژل (بسته به نوع فسفاتاز)، معمولا با CIP (Calf alkaline phosphatase) و یا SAP (Shrimp alkaline phosphatase) تیمار میشود.
۲- استخراج از ژل قطعات برش خورده وکتور و قطعه مورد نظر (Insert)
قطعات ایجاد شدهی حاصل از هضم آنزیمی DNA را با استفاده از الکتروفورز ژل آگارز از یکدیگر جدا کنید. سپس قطعهی مورد نظر خود را بمنظور بازیابی آن از ژل، برش بزنید. داشتن باندهای خوب و واضح و همچنین فضای کافی برای برش زدن باندها در این مرحله مهم است. بنابراین توصیه میشود از یک شانه ژل دارای دانه های بزرگ (که چاهکهای بزرگ در ژل ایجاد میکند) استفاده کنید، بین نمونهها یک چاهک فاصله بگذارید و اجازه دهید تا ژل آهسته ران شود. علاوه بر استفاده از DNAسایز مارکرهای استاندارد، پیشنهاد میشود نمونهی برش نخورده (Uncut) از هر پلاسمید را در کنار نمونهی برش خوردهی آن، روی ژل ران کنید تا در صورتیکه هضم شما مطابق انتظار نباشد، در رفع عیبیابی کمک کند. پس از خالصسازی و استخراج از ژلِ پلاسمید پذیرنده و قطعه مورد نظر (Insert)، غلظت DNA بازیابی شده را تعیین کنید.
۳- اتصال قطعه مورد نظر (Insert) به وکتور
برای اتصال Insert به پلاسمید گیرنده خود از یک لیگاز (DNA ligase) استفاده کنید. در یک واکنش اتصال (Ligation reaction) حدود 100 نانوگرم DNA کل پیشنهاد میشود. ایده آلترین نسبت مولی پلاسمید به Insert، نسبت 1 به 3 است. از آنجایی که تعداد جفت بازهای هر کدام متغیر است، محاسبه این نسبت بر اساس غلظت DNA به تنهایی، دشوار است. بنابراین انجام دو واکنش اتصال برای ساخت هر پلاسمید نوترکیب، با نسبتهای مختلف پلاسمید به Insert توصیه میشود.
همچنین داشتن نمونههای کنترل منفی بطور موازی با واکنش اتصال اصلی (نمونه تست) بسیار مهم است. به عنوان مثال واکنش اتصال DNAی پلاسمیدی بدون حضور Insert، نشان میدهد چه میزان پلاسمید پذیرنده برش نخورده به دلیل حلقوی شدن مجدد آن (Self-ligation) در واکنش وجود داشته است.
۴- ترانسفرم کردن (Transformation)
محصول واکنش اتصال را به باکتری مورد نظر خود ترانسفرم (Transform) یا وارد کنید. دستورالعملهای شرکت سازنده سلولهای مستعد (competent cells) را دنبال کنید. برای اکثر کلونینگهای استاندارد، حدود 1 تا 2 مایکرولیتر از محصول واکنش اتصال را به سلولهای میزبان مستعد مانند DH5alpha یا TOP10 ترانسفرم میکنند. تعداد کلونیهای باکتریایی حاصل از ترانسفرماسیون شما نشانهی خوبی است برای ارزیابی اینکه آیا ترانسفرماسیون شما کار کرده است یا خیر. پلیت حاوی پلاسمید گیرنده و Insert (نمونه تست) باید بطور قابل توجهی دارای کلنیهای بیشتری نسبت به پلیت حاوی پلاسمید گیرنده به تنهایی (نمونه کنترل منفی) باشد.
۵- استخراج پلاسمید نوترکیب
در نهایت باید کلنیهای باکتریایی را بطور جداگانه انتخاب و برداشت کرده و بمنظور استخراج پلاسمید و ارزیابی موفقیتِ واکنش اتصال، در محیط کشت مایع مناسب کشت دهید.
۶- آنالیز پلاسمید نوترکیب
پس از خالصسازی DNA (استخراج پلاسمید)، باید از صحت پلاسمید نوترکیب مطمئن شوید. یکی از روشهای تایید اولیهی وارد شدن Insert در پلاسمید پذیرنده، انجام کلنی PCR با استفاده از پرایمرهای مناسب است. پیشنهاد میشود یک پرایمر روی Insert و پرایمر دیگر روی پلاسمید انتخاب شود. به این تربیب فقط در صورتیکه Insert در جایگاه مورد نظر قرار گرفته باشد، باند مورد نظر مشاهده خواهد شد. همچنین میتوان از آنزیمهای محدود کننده مناسب برای تایید اولیه استفاده کرد. پس از برش با آنزیمهای مناسب، سایز قطعات ایجاد شده در پلاسمید حاوی Insert باید متفاوت از سایز قطعات ایجاد شده در پلاسمید بدون اینسرت (Self-ligated) باشد، و یا از آنزیمی استفاده شود که فقط دارای یک جایگاه برش روی Insert است. در این صورت فقط زمانی پلاسمید خطی میشود که حتما قطعهی مورد نظر در پلاسمید پذیرنده وجود داشته باشد. پس از تایید ابتداییِ ورود Insert در پلاسمید پذیرنده، بمنظور اطمینان قطعی از صحت کار، پلاسمید مورد نظر را تعیین توالی (Sequencing) کنید.
منابع
Traditional Cloning Guide | NEB
Image Sources: Main Pic | Plasmid Map | Traditional Cloning Steps | Traditional Cloning Process
تهیه شده توسط دپارتمان بیولوژی مولکولی شرکت دنازیست آسیا
کیت استخراج پلاسمید (روش ستونی)
۱,۵۱۷,۰۰۰ تومان–۲,۷۰۷,۰۰۰ تومان
{kind=link}
{kind=link}
{kind=link}
19 Comments
امکان داره دو پلازمید که ژن متفاوت وارد یک باکتری بشن؟ اگه آره یا نه چرا؟
ممنون میشم از طریق ایمیل هم پاسخ بدید
با سلام؛
بلی امکان ورود چند پلاسمید وجود دارد ولی اگر oriها در ۲ پلاسمید وارد شده از یک خانواده (گروه) باشند یکی از آنها تکثیر خواهد یافت و دیگری با تکثیر باکتری و در رقابت برای استفاده از دستگاه همانندسازی باکتری حذف خواهد شد. این مکانیسم را INCOMPATIBILITY گویند. فقط یک پلاسمید میتواند همزمان با رشد باکتری تکثیر داشته باشه بنابراین همیشه در یک کلونی باکتری یک نوع پلاسمید هست که استخراج و ارزیابی میکنیم.
ممنونم از توضیحات کامل شما
سلام چرا ۲۰ تا ۳۰ درصد ناقل رشد میکندولی خالی از ژن مورد نظر هستند ممنون میشم جواب بدین
با سلام؛
لطفا سوال خود را کمی واضحتر بپرسید.
سلام اگر محل برش های دو سر قطعه و پلاسمید یکسان نباشد از چه روشی استفاده میکنیم؟
سلام؛
باید گروههای فسفات دو سر وکتور را با استفاده از آلکالین فسفاتاز (آنزیم SAP) قبل از مرحله ligationحذف کنیم.
توجه داشته باشید که در اینصورت، insert شما باید حتما گروه فسفات را داشته باشد تا ligation انجام شود. به ان معنی که اگر insert شما محصول cut آنزیمی از وکتور دیگری است، گروه فسفات را دارد. ولی اگر محصول PCR است، یا باید پرایمرهایی که سفارش میدهید فسفریله باشند؛ و یا اگر پرایمرهای معمولی هستند باید با استفاده از آنزیم PNK که یک کیناز است، گروه فسفات به insert شما اضافه گردد.
مفهوم star activity چیست؟ ممنون میشم توضیح بدید
سلام؛
تحت یک سری شرایط خاص واکنش، آنزیمهای محدود کننده، اختصاصیت خود را از دست میدهند و توالیهای بازی را که متفاوت از سایتهای برشی اصلیشان هستند، برش میزنند. یعنی DNA رو خُرد میکنند به جای اینکه فقط سایت اصلی را برش بزنند.
این پدیده «فعالیت ستارهای یا star activity» نامیده میشود و تقریباً همه آنزیمهای محدود کننده، بسته به آنزیمها، DNA و شرایط واکنش میتوانند فعالیت ستارهای داشته باشند.
یا ممکن است به جای برش دو رشته ی دیاناِی، شکاف در یک رشته ایجاد کنند.
برای جلوگیری از فعالیت ستارهای، توصیه میشود واکنشهای آنزیم را در غلظتهای پایین گلیسرول، pH خنثی و غلظتهای بالاتر نمک انجام دهند. با این حال، این شرایط ممکن است فعالیت آنزیمی کمتری را نیز ایجاد کنند.
زمان هضم طولانی هم میتواند باعث افزایش فعالیت ستارهای شود.
سلام
آیا انتقال DNA به بدن دیگر وجود دارد؟
و در صورت امکان پذیر بودن چه اتفاقی می افتد ؟
سلام؛
لطفا سوال خود را کمی واضحتر بپرسید.
با سلام
امکان دارد از محلول نهایی PCR جهت کلون ژن استفاده کرد و اینکه حتمان باید برش آن از ژل انجام و خالص سازی گردد؟
تشکر
در صورتی برش از روی ژل انجام میشود که سایز محصول بالای ۵۰ جفت باز باشد، در غیر انصورت می توان reaction recover انجام داد (کیتهای ریکاوری DNA دنازیست). بهر حال برای برش آنزیمی محصول PCR و Cloning، باید محصول شما عاری از dNTP ،taq polymerase، بافر، پرایمرها و … باشد.
با سلام واحترام لطفا طریقه محاسبه تعداد کپی نامبر ژن مورد نظر (insert) در یک کلونینگ موفق را توضیح دهید(کیفی و کمی)
با سلام و احترام؛
سوال شما را با یک مثال پاسخ می دهیم.
فرض کنید غلظت insert برش خورده ی مورد نظر 10ng و میزان vector برش خورده ی مورد نظر 5ng است. همچنین سایز insert = 500bp و سایز vector = 7500bp است.
ابتدا با استفاده از سایت زیر نسبت مولی insert به vector را به دست می آوریم؛
https://nebiocalculator.neb.com/#!/ligation
همانطور که میبینید در لینک بالا 3 مقدار از شما خواسته شده است
Insert DNA length = 500bp or 0.5kb
Vector DNA length = 7500bp or 7.5kb
Vector DNA mass = 5ng
بعد از وارد کردن مقادیر بالا، required insert DNA mass با 4 نسبت مختلف نمایان می شود. نسبت 7:1 را در ادامه مورد محاسبه قرار می دهیم. این مقدار یعنی اینکه به ازای 5ng وکتور باید 2.33ng اینسرت استفاده شود. حال اینسرت ما 10ng است. یعنی در هر میکرولیتر 10ng اینسرت داریم پس اگر می خواهیم 2.33ng را وارد واکنش کنیم باید 0.2ul از اینسرت را استفاده کنیم. بنابراین واکنش نهایی Ligation به صورت زیر خواهد بود.
vector = 1ul
insert = 0.2ul
T4 enzyme buffer (10X) = 1ul
T4 enzyme = 0.5
————————–
Total should be 10ul
همانطور که می بینید کل واکنش باید 10ul باشد ولی فعلا 2.7ul است. میتوانیم میزان vector و insert را در یک عدد واحد ضرب کنیم طوریکه حجم نهایی واکنش نزدیک به 10ul گردد. پس:
vector = 1ul X 7 = 7ul
insert = 0.2ul X 7 = 1.4ul
T4 enzyme buffer (10X) = 1ul
T4 enzyme = 0.5
————————–
Total should be 10ul
یعنی در این واکنش ligation می بایست 7 میکرولیتر وکتور، 1.4 میکرولیتر اینسرت، 1 میکرولیتر بافر T4 لیگاز و 0.5 میکرولیتر T4 لیگاز استفاده گردد.
سلام وقتتون به خیر باشه،من سوالی از خدمتتون دارم که تا قبل از هفته دیگه باید جوابش رو بدونم ،ممنون میشم راهنماییم کنید
دو تا سوپانسیون با غلظت های مساوی از پلاسمید و ژنوم جو داریم (میدونیم یکیش ژنومش کوچیکه ،یکیش بزرگ)از کدومشون باید dna بیشتری برداریم تا در نهایت تعداد نسخه های مساوی ازشون تو pcrداشته باشیم ،ممنون میشم اینجا یا در ایمیل جوابم رو بدید
با تشکر
با سلام و احترام؛ پاسخ هر سه سوال شما:
وارد سایت https://www.technologynetworks.com/tn/tools/copynumbercalculator شوید.
در قسمت Length of Template (bp) سایز DNA ژنومی یا پلاسمیدی را با واحد base pair وارد کنید.
در قسمت DNA Concentration (ng/μL) غلظت DNA ژنومی یا پلاسمیدی را با واحد ng/ μL وارد کنید.
بعد از فشردن tab سبز رنگ Calculate نتیجه به صورت Number of Copies per μL ظاهر میشود.
حال می توانید با بستن تناسب، تعداد کپی های DNA ژنومی و پلاسمیدی را همسان سازی کنید.
موفق باشید.
سوالم اینه که اگر dnaالگو ما بزرگ تر باشه باید مقدار بیشتری استخراج کنیم با dnaالگو ما کوچیک تر باشه؟
برای اینکه تعداد نسخه های مساوی در نهایت داشته باشیم باید از پلاسمید dna بیشتری برداریم یاژنوم جو و چرا؟
ممنون میشم جوابم بدید ،با تشکر