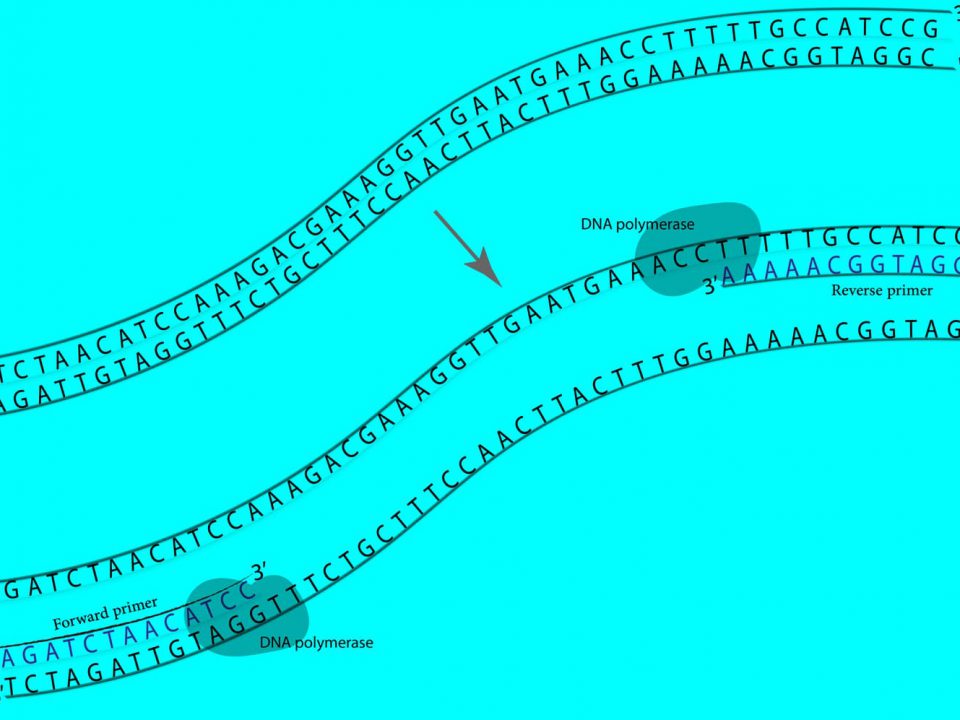
راهنمای طراحی پرایمرهای PCR
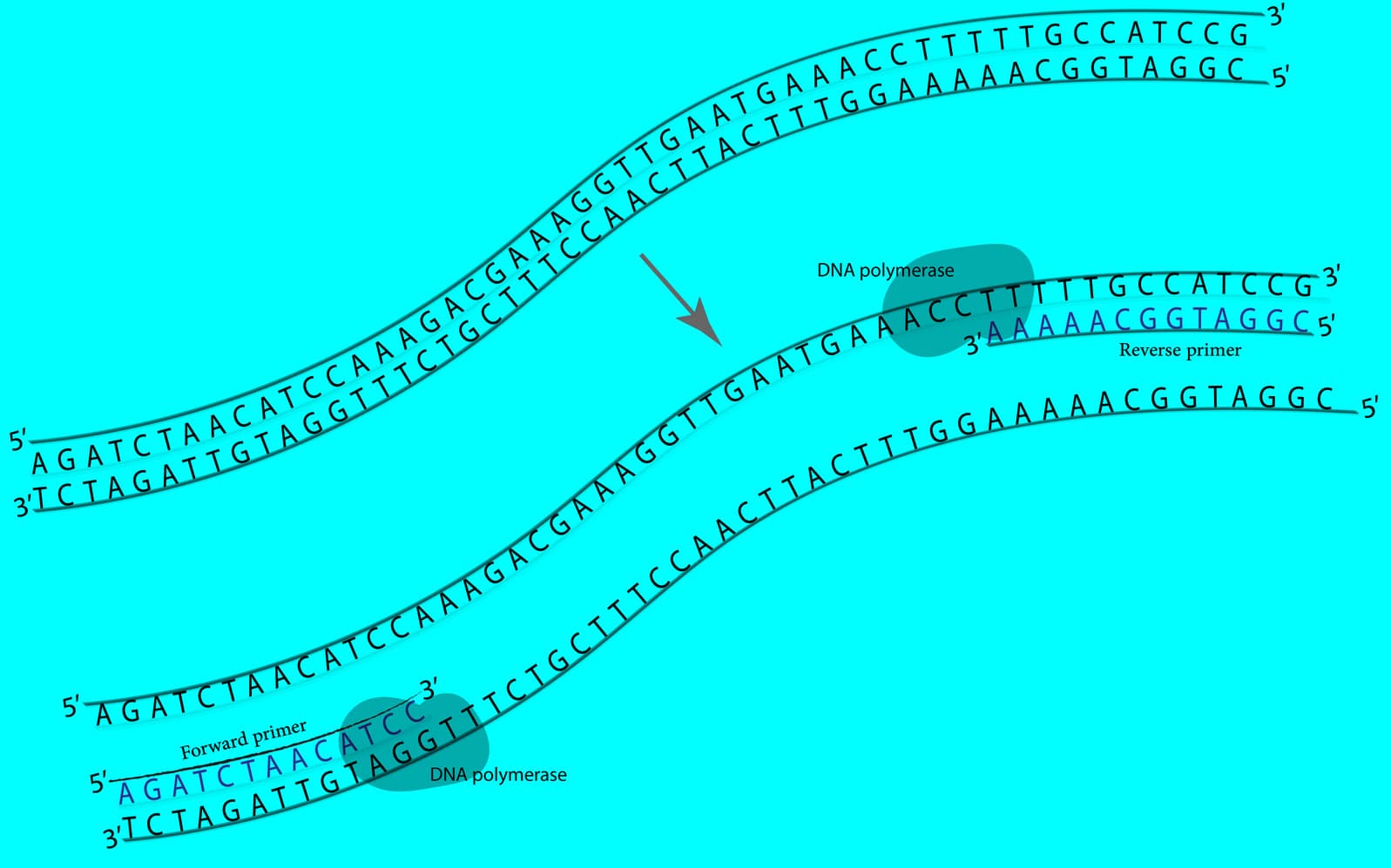
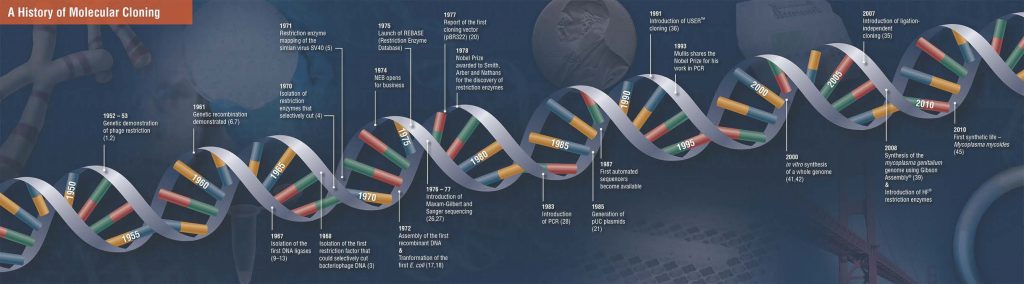
7 خرداد 1399کلونینگ مولکولی
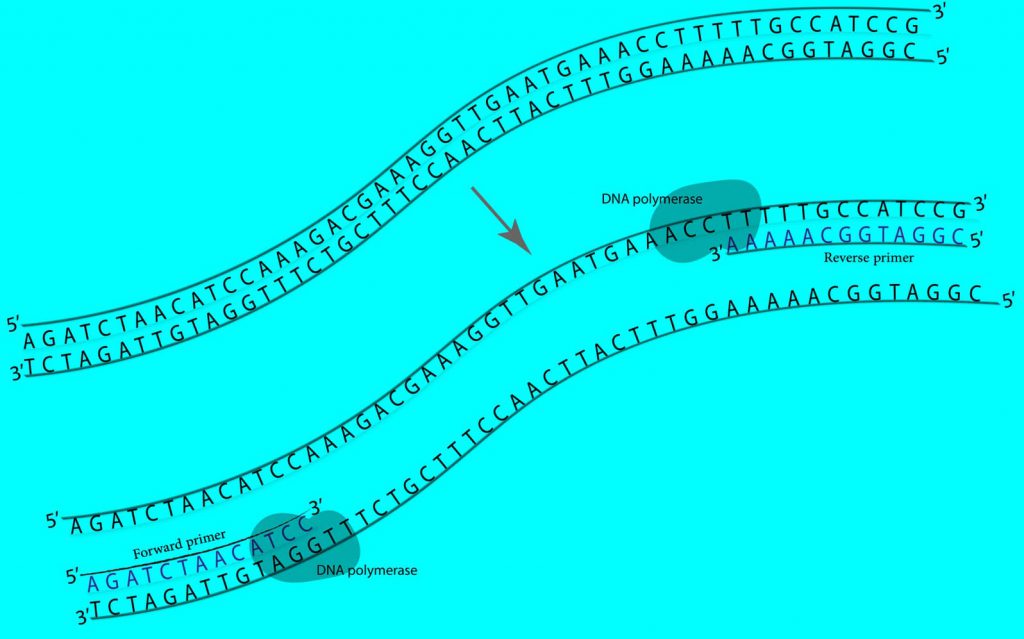
6 تیر 1399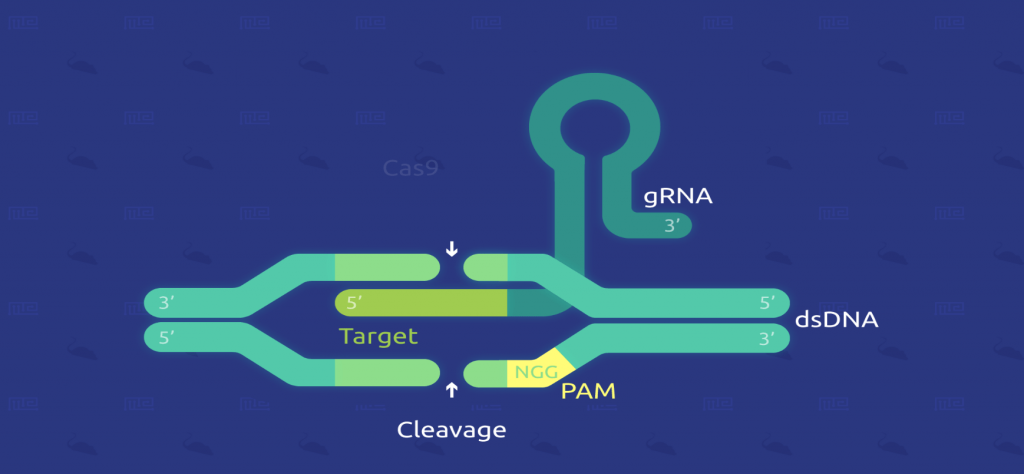
ویرایش ژنوم
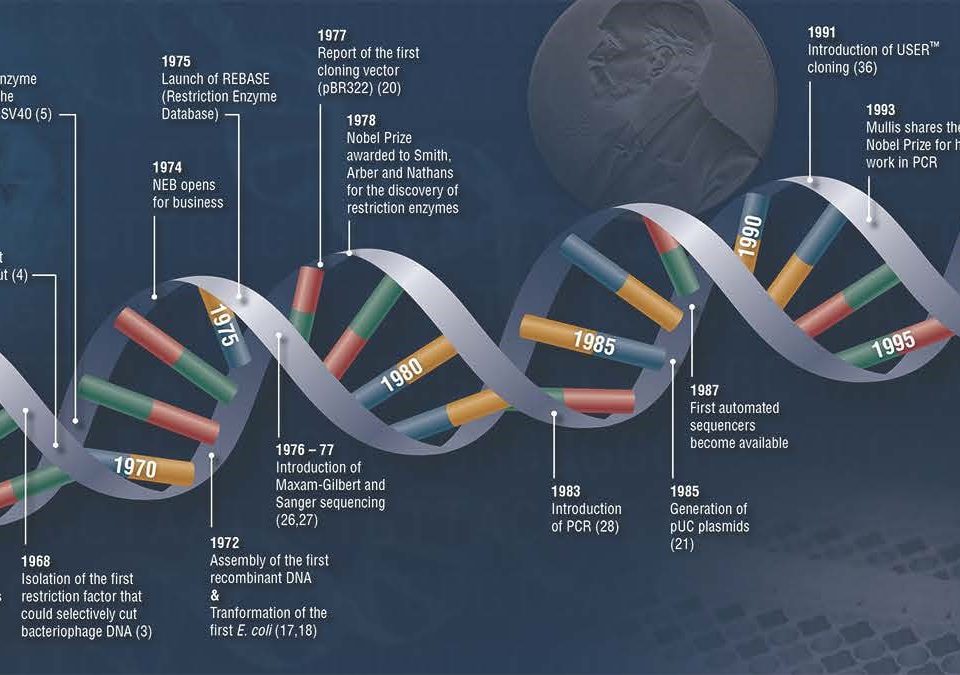
ویرایش ژنوم (Genome editing) این امکان را فراهم میکند تا با توسعه ابزارها، تغییرات دقیق و هدفمندی در ژنوم سلولهای زنده ایجاد شود. در اواخر دههی ۱۹۷۰، کشف آنزیمهای محدود کننده که بهطور طبیعی از باکتریها در برابر فاژها (Phages) محافظت میکنند، نقطه عطفی در ظهور DNA نوترکیب (Recombinant DNA) بود. برای اولین بار با استفاده از این آنزیمها، دانشمندان توانایی دستکاری DNA در لولههای آزمایش را پیدا کردند.
اگرچه چنین تلاشهایی، اکتشافات جدیدی را در زیست شناسی مولکولی و ژنتیک به دنبال داشت، اما توانایی تغییر دقیق و هدفمند DNA در سلولهای زندهی یوکاریوتی چند دهه بعد ایجاد شد. در اواخر دهه ۱۹۸۰ دانشمندان با استفاده از فرایند نوترکیبی همولوگ (یا Homologous recombination)، توانستند DNA خارجی را در ژنوم سلولهای پستانداران وارد کنند.
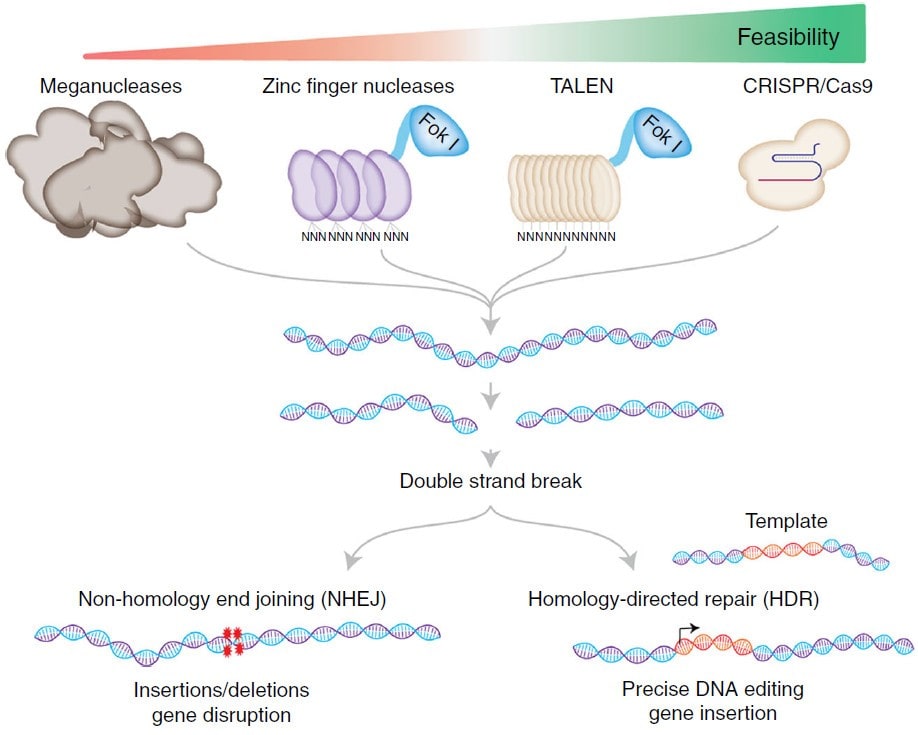
با وجود موفقیت این روش در دستکاری هدفمند ژنوم، نوترکیبی همولوگ دارای محدودیتهایی بود. به دنبال تلاش برای غلبه بر این محدویتها، محققان دریافتند که ایجاد شکست دو رشتهای (Doublestrand Break; DSB) در محل هدف، احتمال ادغام هدفمند ژن (Targeted gene integration) را افزایش میدهد. در این راستا کشف و استفاده از مگانوکلئازها (Meganucleases)، و همچنین اتصال اندونوکلئازهایی مانند Fok1 به پروتئینهای Zinc finger یوکاریوتی (Zinc finger nucleases; ZFN) و یا به پروتئینهای TALE (یا Transcription activator-like effector nucleases; TALEN) عصر جدیدی را در هدف قرار دادن و ویرایش ژنوم آغاز کرد.
اگرچه کشف مگانوکلئازهای مصنوعیِ طراحیشده توسط ZFNها و TALENها، کارایی ویرایش ژنوم را به میزان زیادی افزایش داد، اما هدف قرار دادن سایتهای مختلف در ژنوم، نیازمند طراحی مجدد یا مهندسی مجدد مجموعه جدیدی از پروتئینها بود.
دشواری کلونینگ و مهندسی پروتئین ZFNها و TALENها، تا حدی مانع از استفاده گسترده این ابزارها توسط محققین شد. از این منظر، کشف CRISPR به دلیل سهولت و انعطافپذیری بیشتر، انقلابی را در ویرایش هدفمند ژنوم ایجاد کرد (تصویر ۱).
مگانوکلئازها، آنزیمهای محدودکننده مهندسیشدهای هستند که توالیهای طولانی DNA را تشخیص میدهند. هر نوکلئاز Zinc finger (یا ZFN) یک کد سهتایی DNA (یعنی ۳ جفت باز)، در حالی که هر TALE میتواند یک جفت باز را شناسایی کند. بر خلاف شناسایی DNA بر مبنای پروتئینِ در ZFNها و TALENها، مکانیسم هدف قرار دادن ناحیهی خاصی از ژنوم در سیستم CRISPR، بر اساس جفتشدن سادهی بازهای RNA-DNA و توالی PAM است.
تمامی این ابزاها باعث ایجاد شکست دورشتهای DNA میشوند، سپس با اتصال انتهای غیرهمولوگ مستعد خطا (Error-prone non-homology end joining; NHEJ) و یا ترمیم هدایت شدهی هوموگ (Homology-directed repair; HDR)، این شکست ایجاد شده در ژنوم ترمیم میگردد. در حالیکه NHEJ منجر به حذف و یا اضافه شدن (Insertions and/or deletions; Indel) تصادفی بازها در محل موردنظر و اختلال در ژن مربوطه میشود، HDR میتواند با وارد کردن یک الگوی خاص DNA (تکرشته یا دورشته) در سایت موردنظر، برای ویرایش دقیق ژن مورد استفاده قرار گیرد.
CRISPR چیست؟
عملکرد ژنهای CRISPR (یا Clustered Regularly Interspaced Short Palindromic Repeats) و ژنهای مرتبط با CRISPR (یا CRISPR-associated; Cas) در ایمنی اکتسابیِ باکتریها و آرکیا (archaea) ضروری است و باعث میشود این ارگانیسمها بتوانند به مواد ژنتیکی مهاجم، پاسخ دهند و آنها را از بین ببرند. این تکرارها برای اولین بار در دهه ۱۹۸۰ در باکتری E. coli کشف شدند، اما عملکرد آنها تا سال ۲۰۰۷ ناشناخته ماند تا اینکه Barrangou و همکارانش نشان دادند باکتری S. thermophilus با ادغام یک قطعه ژنومی از یک ویروس مهاجم به داخل لوکوس CRISPR خودش، میتواند در برابر باکتریوفاژ مقاوم شود.
سه کلاس از سیستم CRISPR شناسایی شده است که نوع ۲ (Class II) بیشتر از بقیه مورد مطالعه قرار گرفته است. در این نوع، DNA مهاجمِ ویروسها یا پلاسمیدها به قطعات کوچک بریده میشود و در میان مجموعهای از تکرارهای کوتاه (در حدود ۲۰ جفت باز)، در یک لوکوس CRISPR باکتری گنجانده میشوند. سپس لوکوسها رونویسی میشوند و رونوشتها پردازش شده و RNAهای کوچکی (CRISPR RNA; crRNA) را تولید میکنند که منجر به هدایت اندونوکلئازهای عمل کننده (Effector endonucleases) برای هدف قرار دادن DNA مهاجم، بر اساس توالی مکملشان میشود (تصویر ۲).
در سیستمهای CRISPR نوع ۲ (Type II CRISPR systems) مشخص شده است که پروتئین Casی به نام Cas9 (یا Csn1) دارای نقش کلیدی است و باعث خاموش کردن ژن (Gene silencing) در این سیستمها میشود. Cas9، در پردازش crRNAها شرکت میکند و مسئول تخریب DNAی هدف است. عملکرد Cas9 در هر دو مرحله، به دلیل حضور دو دامینِ شبه نوکلئازی به نامهای RuvC (یا RuvC-like nuclease domain) و HNH (یا HNH-like nuclease domain) است.
بمنظور تشخیص اختصاصی یک سایت (Site-specific DNA recognition) و برش DNA، باید یک کمپلکس پروتئین- RNA، شامل پروتئین Cas9، یک crRNA و یک tracrRNA (یا trRNA)، که تا اندازهای مکملِ crRNA است، تشکیل شود. tracrRNA برای بلوغ crRNA از یک رونوشت اولیهی کدکنندهی چندین پیش crRNA (یا pre-crRNAs) مورد نیاز است. این اتفاق در حضور RNase III و Cas9 رخ میدهد.
در حین تخریب DNAی هدف، دُمینهای شبه نوکلئازی HNH و RuvC، هر دورشتهی DNA را برش میزنند و باعث ایجاد شکستهای دورشتهای (Double-stranded breaks; DSBs) در سایتهای تعیین شده توسط یک توالی ۲۰ نوکلئوتیدی در داخل رونوشت crRNA مربوطه، میشوند. دمین HNH رشتهی مکمل و دمین RuvC رشتهی غیر مکمل را برش میزند.
فعالیت اندونوکلئازیِ دو رشتهای Cas9 همچنین مستلزم این است که یک توالی کوتاه (۲ تا ۵ نوکلئوتیدی) حفاظتشده به نام Protospacer-associated motif (یا PAM)، بلافاصله در انتهای ۳ پرایمِ (‘3) توالی مکملِ crRNA (در DNA هدف) وجود داشته باشد. در حقیقت حتی توالیهای کاملاً مکمل نیز در غیاب این توالی PAM، توسط Cas9-RNA نادیده گرفته میشوند (تصویر ۲).
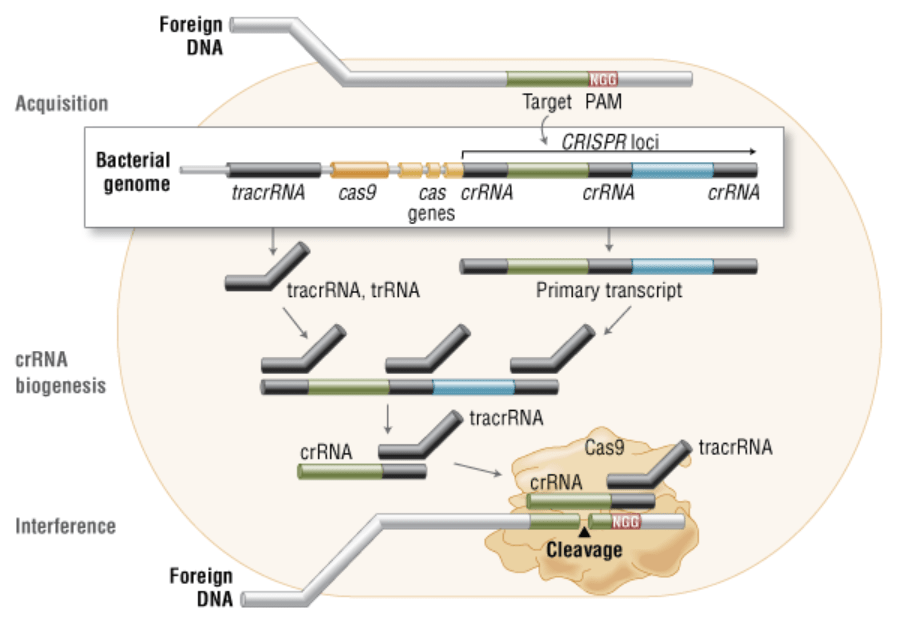
در ایمنی اکتسابی، DNA خارجی (مهاجم) در ژنوم باکتریایی، در لوکوس CRISPR گنجانده میشود. سپس لوکوس CRISPR رونویسی و بصورت crRNAهای کوچک پردازش میشود. اندونوکلئاز Cas9 با یک crRNA و tracrRNA مجزا تشکیل یک کمپلکسی را میدهد که میتواند DNA خارجیِ حاوی یک توالی ۲۰ نوکلئوتیدیِ مکمل با crRNA، در مجاورت توالی PAM را برش بزند (شکل با مقیاس رسم نشده است).
CRISPR و Cas9 به عنوان ابزاری جدید در زیست شناسی مولکولی
سادگی نوکلئاز CRISPR نوع ۲، تنها با سه جزء مورد نیاز (Cas9 به همراه crRNA و tracrRNA)، این سیستم را به ابزاری پرکاربرد برای ویرایش ژنوم تبدیل کرده است. این پتانسیل در سال ۲۰۱۲ توسط آزمایشگاههای Doudna و Charpentier تحقق یافت. بر اساس سیستم CRISPR نوع ۲ که در بالا توضیح داده شد، محققین با ترکیب trRNA و crRNA بهصورت یک RNAی راهنمای سنتز شدهی مصنوعی (single guide RNA; sgRNA)، یک سیستم دو جزئی ساده تولید کردند. در این سیستم ساده، Cas9 توسط یک sgRNA به ناحیهی موردنظر هدایت، و منجر به تغییرات هدفمند ژنوم میشود.
انواع مختلف Cas 9
تا به امروز ، سه نوع مختلف از نوکلئاز Cas9 در پروتکلهای ویرایش ژنوم مورد استفاده قرار گرفته است:
۱- نوع وحشی Cas 9
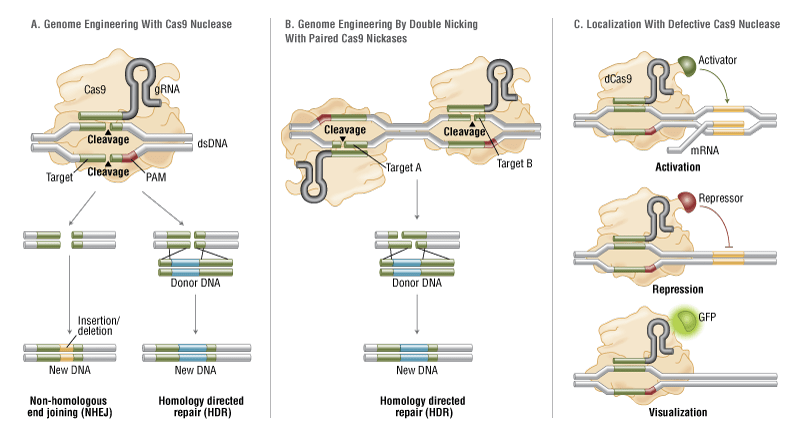
نوع وحشی Cas9 (یا Wild-type Cas9 nuclease) که میتواند بهطور اختصاصی DNA را بصورت دورشتهای برش بزند و در نتیجه منجر به فعالسازی ماشینهای ترمیم شکستهای دورشتهای (DSBs) داخل سلولی شود. در صورت عدم وجود الگوی ترمیم همولوگ، ترمیم از مسیر اتصال انتهای غیرهمولوگ (NHEJ) پیش میرود و در نتیجهی ورود و یا حذف بازها (Indels) در محل موردنظر، منجر به اختلال در توالی هدف میشود. از طرف دیگر، با فراهم کردن یک الگوی ترمیم همولوگ و بهرهمندی از مسیر ترمیم هومولوژی (HDR)، میتوان جهشهای دقیقی را در محل موردنظر، از طریق خارج کردن (Knock-out) و یا وارد کردن (knock-in) یک توالی خاص انجام داد (تصویر ۳، قسمت A).
۲- نوع جهشیافتهی Cas 9
فرم جهشیافتهی Cas9 (یا Mutated Cas9 or nickase Cas9; nCas9) که میتواند یک شکاف تکرشتهای (Single-strand nick) در محل موردنظر ایجاد کند. بنابراین مسیر ترمیم NHEJ فعال نشده و احتمال جهشهای Indel کاهش مییابد. بهمنظور برش دورشتهای و ترمیم HDR، باید با طراحی دو sgRNA ،دو شکاف مجاور در محل موردنظر ایجاد کرد (تصویر ۳، قسمت B).
۳- نوع Cas 9 فاقد فعالیت نوکلئازی
فرم Cas9 فاقد فعالیت نوکلئازی (یا Nuclease-deficient Cas9; dCas9) که با موتاسیون در دمینهای نوکلئازی آن قادر به برش DNA نیست، اما هنوز میتواند با هدایت sgRNA به محل موردنظر متصل شود. بنابراین دمینهای عملکنندهی مختلف (فعالکنندههای رونویسی، سرکوبکنندهها و پروتئینهای فلورسنت) را میتوان با اتصال به dCas9 به محل موردنظر هدایت کرد (تصویر ۳، قسمت C).
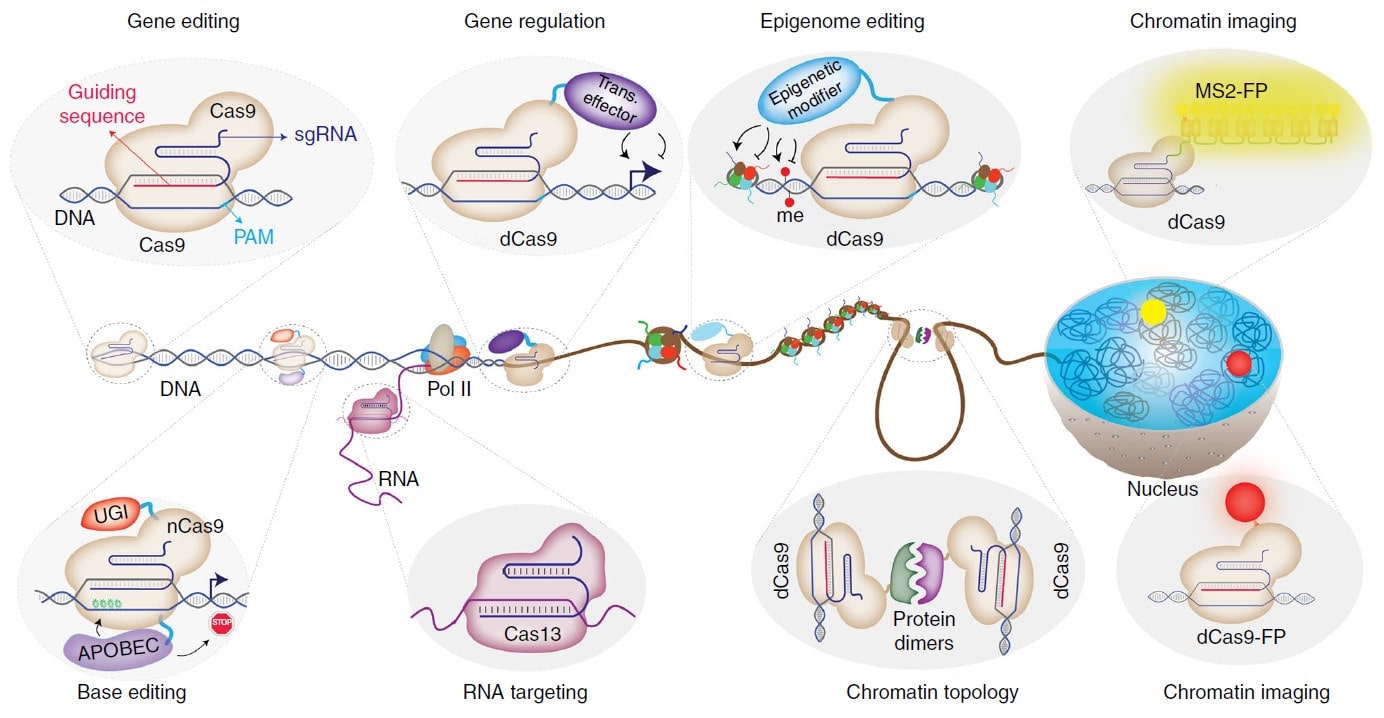
پس از معرفی اولیه در سال ۲۰۱۲، CRISPR به ابزاری ضروری در تحقیقات بیولوژیکی تبدیل شده است. امروزه قابلیت برنامهریزیشدهی آنزیم Cas 9، تحقیقات متنوع در زمینههای پزشکی، بیوتکنولوژی و کشاورزی را متحول کرده است. CRISPR-Cas9 دیگر فقط ابزاری برای ویرایش ژن نیست. زمینههای کاربردی Cas 9 غیرفعال (dCas9)، شامل تنظیم ژن (Gene regulation)، ویرایش اپیژنتیک (Epigenetic editing)، مهندسی کروماتین (Chromatin engineering)، تصویربرداری کروماتین (Chromatin imaging) و غیره، اکنون از قابلیت ویرایش ژن Cas9 نوع وحشی فراتر رفته است (تصویر ۴).
چگونه از CRISPR در تحقیقات خود استفاده کنیم؟
با بیان همزمان اندونوکلئاز Cas9 یا Cas12a (که به عنوان Cpf1 نیز شناخته میشود) و یک gRNA اختصاصی برای ژن هدف، میتوانید از CRISPR برای ایجاد یک دستکاری در ژنوم استفاده کنید. هدف ژنومی میتواند هر توالی DNA ژنومی در حدود ۲۰ نوکلئوتید باشد، به شرط آنکه دارای دو ویژگی زیر باشد:
۱- این توالی در مقایسه با بقیه ژنوم منحصر به فرد باشد.
۲- توالی هدف بلافاصله در مجاورت PAM (یا Protospacer Adjacent Motif) قرار گرفته باشد.
توالی PAM به عنوان یک سیگنال اتصالی برای Cas9 عمل میکند، اما توالی دقیق آن به پروتئینِ Casی بستگی دارد که شما در آزمایش خود استفاده میکنید. به عنوان مثال توالی PAM برای Cas9، توالی NGG است (N میتواند هر کدام از چهار نوکلئوتید A، T، C یا G باشد)، درحالیکه توالی PAM برای Cpf1، توالی TTTV است (V میتواند A، C، G باشد).
قدم نخست: هدف خود را تعیین کنید
اولین قدم برای استفاده از CRISPR برای دستکاری ژنتیکی، اینست که هدف خود را مشخص کنید، آیا میخواهید:
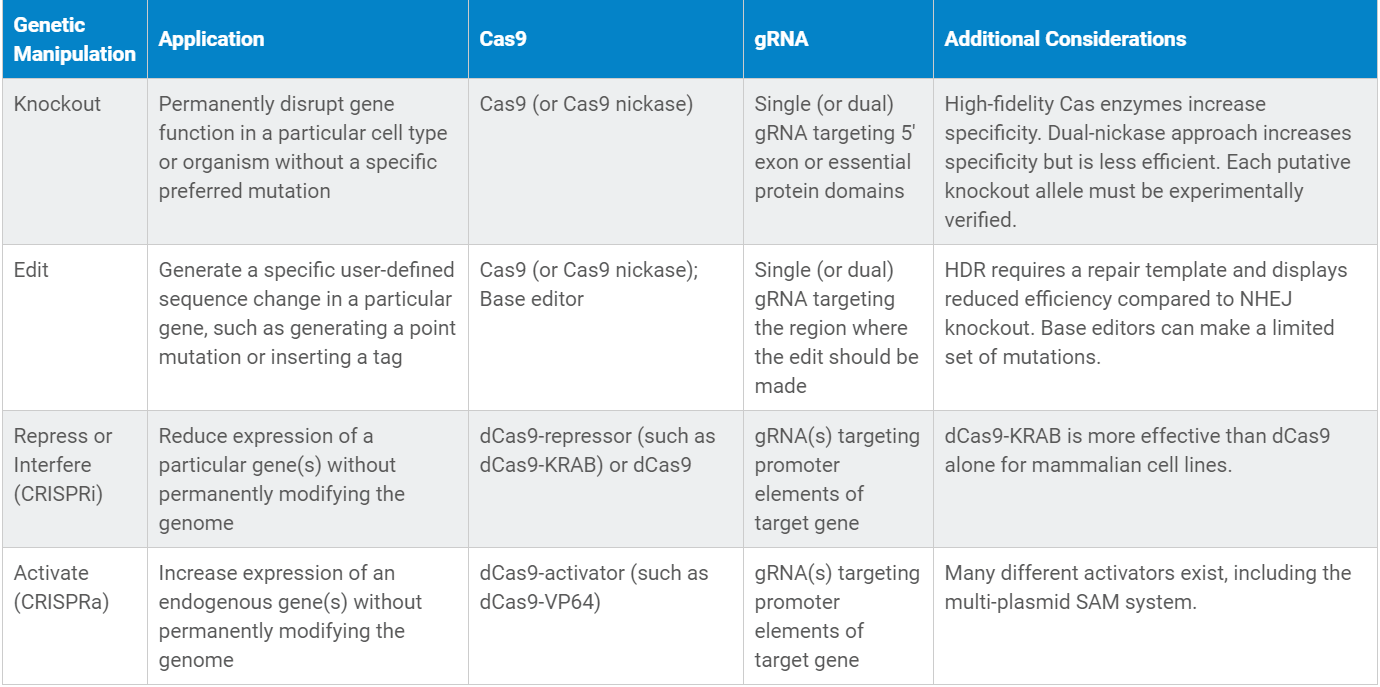
- بیان ژن یا عملکرد آن را بهطور کامل یا دائمی از بین ببرید (Knockout کنید)؟
- جهش خاصی در یک آلل از یک ژن (جهش نقطهای) ایجاد کنید؟
- بیان یک ژن هدف را افزایش یا کاهش دهید؟
- یک ژن (مثلا یک ژن گزارشگر) یا توالی خاصی را در ناحیهی مشخصی از ژنوم وارد کنید (Knockin کنید)؟
زمانیکه درک روشنی از هدف آزمایشی خود داشته باشید، آمادگی لازم را برای هدایت عوامل مختلفی که برای آزمایش خاص شما در دسترس است، خواهید داشت.
قدم دوم: دستکاری ژنتیکی موردنظر خود را انتخاب کنید
دستکاریهای ژنتیکی مختلف، به اجزای مختلف CRISPR احتیاج دارد. با انتخاب یک دستکاری ژنتیکی خاص میتوانید تعیین کنید که کدام پلاسمید CRISPR برای آزمایش خاص شما مناسب است. اطمینان حاصل کنید که آیا این پلاسمید برای انجام آزمایش در ارگانیسم مدنظر شما موجود هست یا خیر. برای کاربرد خاص شما ممکن است پلاسمید مناسب وجود نداشته باشد. در چنین مواردی ممکن است لازم باشد یک پلاسمید موجود را متناسب با نیاز خود تغییر دهید و اصلاح کنید.
قدم سوم: سیستم بیانی مناسبی را انتخاب کنید
برای استفاده از CRISPR، به Cas9 و یک gRNA نیاز دارید که در سلولهای هدف شما بیان شوند. برای انواع سلولهایی که به راحتی ترانسفکت میشوند (مانند سلولهای HEK293)، مواد ترانسفکشنِ استاندارد برای بیان CRISPR ممکن است کافی باشد. برای سلولهایی که به سختی ترانسفکت میشوند (مانند سلولهای کشت اولیه)، تحویل ویروسیِ پلاسمید CRISPR ترجیح داده میشود. در مواردی که ویرایش خارج از هدف (Off-target editing) نگران کننده است، استفاده از کمپلکس ریبونوکلوپروتئین Cas9-gRNA (یا Cas9-gRNA ribonucleoprotein; RNP)، به دلیل بیانِ گذرای Cas9 توصیه میشود.

قدم چهارم: توالی هدف موردنظر را انتخاب و gRNA خود را طراحی کنید
پس از انتخاب پلاسمید CRISPR و روش تحویل مناسب، باید توالی هدف خود را انتخاب کنید و برای این ناحیه، gRNA مناسبی طراحی کنید.
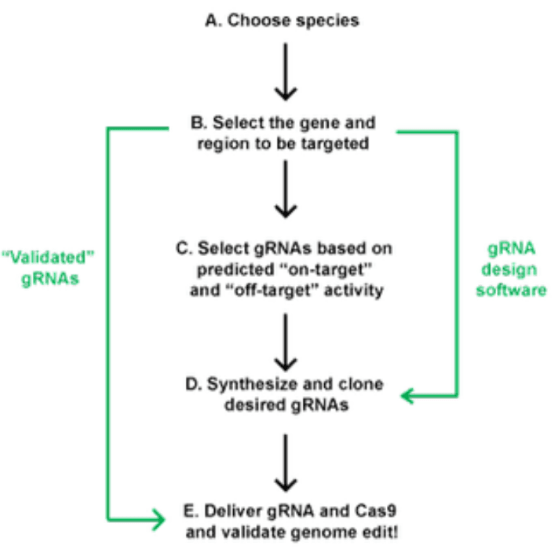
قدم پنجم: رده سلولی / ارگانیسم و توالی ژنومی خود را بشناسید
در صورت امکان، قبل از طراحی gRNA، ناحیهی ژنومی خود را که قصد دارید دستکاری کنید، تعیین توالی نمایید. زیرا تفاوت توالی بین gRNA و DNA هدف ممکن است منجر به کاهش کارایی ویرایش ژنوم شود. تعداد آللهای مربوط به هر ژن بسته به نوع سلول یا ارگانیسم خاص ممکن است متفاوت باشد، که احتمال دارد کارایی knockout و یا knockin سیستم CRISPR را تحت تأثیر قرار دهد.
قدم ششم: ژن و عنصر ژنتیکی مناسبی را برای دستکاری انتخاب کنید
برای دستکاری یک ژن معین با استفاده از CRISPR، باید توالی ژن موردنظر خود را شناسایی کنید. با این حال ناحیهی دقیق ژن موردنظر شما به کاربرد خاص شما بستگی دارد. مثلا:
- برای فعالسازی یا سرکوب ژنِ هدف با استفاده از فعالکنندهها (dCas9-activators) یا سرکوبکنندهها (dCas9-repressors)، بایستی gRNA های لازم باید برای ناحیه پروموتری ژن طراحی شوند.
- برای knockout های ژنتیکی، gRNA ها معمولاً باید اگزونهایی که در ناحیه ۵ پرایم (‘5)، بطور دائمی بیان میشوند را هدف قرار دهند. باید اگزونهای نزدیک انتهای N مورد هدف قرار گیرند زیرا جهشهای تغییر قالب (Frameshift) در این ناحیه، احتمال تولید یک محصول پروتئینی غیرفعال را افزایش میدهند.
- همچنین gRNAها میتوانند برای هدف قرار دادن اگزونهایی که دمینهای ضروری پروتئین را کد میکنند، طراحی شوند. بنابراین حتی اگر تغییر قالب اتفاق نیافتد، عملکرد پروتئین تغییر خواهد کرد.
- برای آزمایشهای ویرایش ژن با استفاده از HDR، ضروری است که توالی هدف بسیار نزدیک به محل ویرایش موردنظر باشد، در حالت ایده آل در فاصلهای کمتر از ۱۰ جفت باز. در این حالت لازم است مکان دقیقی را که ویرایش باید در آن رخ دهد، شناسایی کنید و یک توالیِ هدف در نزدیکی آن انتخاب کنید.
قدم هفتم: gRNAها را بر اساس فعالیت پیشبینی شده روی هدف (On-target) و خارج از هدف (Off-target) انتخاب کنید
توالی PAM، برای عملکرد Cas9 در اتصال به DNAی هدف ضروری است. بنابراین میتوان با شناسایی تمام توالیهای PAMی که در ناحیه ژنومی میتوانند مورد هدف قرار گیرند، شروع کرد. اگر توالی PAM برای آنزیم انتخابی شما، در توالی موردنظر وجود نداشته باشد، میتوانید از دیگر آنزیمهای جایگزین Cas (انواع Cas و توالی PAM آنها در جدول زیر نشان داده شده است) استفاده کنید. پس از شناسایی توالی PAM، سایت هدف مناسبی را انتخابِ کنید. باید سایت هدفی انتخاب شود که کارآمدترین نتیجه در ایجاد برش روی هدف (On-target) حاصل گردد.

توالی gRNA باید با لوکوس موردنظر مطابقت داشته باشد، اما اطمینان از اینکه این توالی gRNA، با سایر سایتهای دیگر در ژنوم مطابقت نداشته باشد نیز بسیار مهم است. در یک شرایط ایدهآل، توالی gRNA شما، هومولوژی کامل با هدف شما دارد و با هیچ جای دیگری از ژنوم هومولوژی ندارد. اما در شرایط واقعی اینطور نیست و توالی gRNA تا حدودی با سایر سایتهای دیگر در سراسر ژنوم، هومولوژی جزئی خواهد داشت. به این سایتها، نواحی خارج از هدف (Off-target) گفته میشود و باید در زمان طراحی gRNA مورد بررسی قرار گیرند.
بهطور کلی سایتهای Off-target، زمانیکه در نزدیکی توالی PAM، عدم تطابق (Mismatches) بازها وجود داشته باشد، به همان اندازهی سایتهای On-target با همومولوژی کامل در نزدیکی PAM، کارآمد نیستند. بنابراین gRNAهای بدون هومولوژی یا آنهایی که دارای عدم تطابق نزدیک به توالی PAM هستند، دارای بالاترین اختصاصیت خواهند بود. همچنین شما میتوانید برای افزایش اختصاصیت، از آنزیم Casِ High-fidelity (یا High-fidelity Cas enzyme) استفاده کنید.
علاوه بر فعالیت Off-target، در نظر گرفتن عواملی که باعث به حداکثر رساندن برش توالی هدفِ موردنظر یا فعالیت On-target میشوند نیز مهم است. دو توالی gRNA با ۱۰۰٪ همولوژی با اهداف DNA، ممکن است کارايی یکسانی در برش محل هدف نداشته باشند. در حقیقت، بسته به نوکلئوتیدهای خاص موجود در توالی هدف انتخابی، راندمان برش ممکن است افزایش یا کاهش یابد. به عنوان مثال، توالیهای gRNA حاوی نوکلئوتید G در موقعیت ۲۰ (۱ جفت باز در بالادست PAM) ممکن است مؤثرتر از gRNAهای حاوی نوکلئوتید C در همان موقعیت باشند (علیرغم داشتن تطابق کامل با توالی هدف).
بسیاری از برنامههای طراحی gRNA میتوانند توالی PAM و هدف بالقوه را پیدا کرده و gRNAهای مرتبط را براساس فعالیتهای پیشبینی شده Off-target و On-target رتبه بندی کنند (به نرم افزار طراحی gRNA لیست شده در ذیل مراجعه کنید).
- http://chopchop.cbu.uib.no
- https://eu.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM
- http://crispr.dbcls.jp
- https://www.benchling.com/crispr
- http://crispor.tefor.net
- http://www.e-crisp.org/E-CRISP/designcrispr.html
- https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design
- http://grna.ctegd.uga.edu
قدم هشتم: سنتز و کلون کردن gRNA های موردنظر
پس از انتخاب توالی هدف خود، باید الیگونوکلئوتید gRNA خود را طراحی کرده و این الیگوها را در وکتور موردنظر خود کلون کنید. در بسیاری از موارد، این الیگوها، سنتز، Anneal شده و در پلاسمیدهای حاوی جایگاه برای gRNA، با استفاده از کلونینگ استاندارد (واکنش هضم آنزیمی- اتصال) وارد میشوند. با اینحال استراتژی کلونینگ دقیق، به وکتورِ gRNAای که انتخاب کردهاید، بستگی دارد، بنابراین بهتر است پروتکل مرتبط با پلاسمید خاص موردنظر خود را دنبال کنید.
قدم نهم: انتقال Cas9 و gRNA به سلول
روش انتقال مناسبی را انتخاب کنید که با سیستم آزمایشگاهی شما سازگار باشد. راندمان CRISPR براساس روش تحویل و نوع سلول میتواند متفاوت باشد. قبل از شروع آزمایش خود، بهتر است شرایط ترانسکفشن خود را بهینه کنید.
قدم دهم: ارزیابی ویرایش ژنتیکی
پس از موفقیت انتقال آنزیم Cas و gRNA به سلولهای هدف، وقت آن است که ویرایش ژنوم خود را تأیید کنید. ویرایش CRISPR، چندین ژنوتیپ مختلف را در جمعیت سلولی ترانسفکت شده ایجاد میکند. بعضي از سلولها ممكن است ۱) به علت عدم وجود gRNA و يا عدم بيان Cas9، و ۲) فقدان برش موثر در سلولهای بیانکنندهی gRNA و Cas9، دارای ژنوتیپ وحشی باشند.
سلولهای ویرایش شده ممکن است برای ویرایش در محل موردنظر شما هموزیگوت یا هتروزیگوت باشند. بهعلاوه در سلولهای حاوی دو آلل جهشیافته، هر آلل جهشیافته ممکن است با توجه به ماهیت مستعد خطای NHEJ متفاوت باشد. در آزمایشهای ویرایش ژن HDR، بیشتر آللهای جهشیافته حاوی ویرایش دلخواه نیستند، زیرا درصد زیادی از DSBها هنوز توسط NHEJ ترمیم میشوند.
چگونه تشخیص میدهید ویرایش موردنظر شما رخ داده است؟
روش دقیقِ لازم برای تأیید ویرایش شما، به دستکاری خاص شما بستگی دارد. با اینحال چندین روش متداول برای تأیید اینکه سلولهای شما حاوی ویرایش موردنظر هستند، وجود دارد:
۱- Mismatch-cleavage assay (برای DSBهای ترمیم شده با NHEJ)
این روش، خوانش نیمه کمی از درصد آللهایی را که در یک جمعیت سلولیِ مختلط جهشیافتهاند را فراهم میکند. ناحیهی موردنظر با PCR تکثیر میشود، محصولات PCR دناتوره-رناتوره میشوند، سپس با نوکلئازی که هترودوبکسهای DNA را برش میزند تیمار میشوند، و در آخر برای شناسایی قطعات DNA روی ژل آگارز برده میشوند (T7 Endonuclease Assay).
۲- PCR و هضم با آنزیمهای محدودکننده (برای DSBهای ترمیم شده با HDR)
این روش برای ویرایشهای نوکلئوتیدی کوچک که یک سایت شناسایی جدید برای یک آنزیم محدودکننده را ایجاد میکنند، مناسب است. منطقه موردنظر با PCR تکثیر شده، با آنزیم محدود کننده مناسب هضم میشود و برای شناسایی قطعات DNA بر روی ژل آگارز برده میشود.
۳- تکثیر با PCR و الکتروفورز ژل (برای HDR یا NHEJ)
برای حذفهای (Deletions) بزرگ یا قطعات گنجانده شده (Insertions) بزرگ در ژنوم، ناحیه موردنظر را میتوان با استفاده از پرایمرهایی که (1) در دوطرف ناحیه موردنظر (deletionها و یا insertion) یا (2) روی مرز ژنوم- insert طراحی شدهاند (فقط برای Insertionها)، با PCR تکثیر کرد. سپس محصول PCR روی ژل آگارز برده میشود تا مشخص شود، آیا ویرایش موفقیت آمیز بوده است یا خیر.
۴- تکثیر باPCR ، subclone کردن در یک پلاسمید و توالی یابی Sanger (برای HDR یا NHEJ)
این روش، ارزیابی نیمه کمی از فرکانس دستکاری و توالی دقیق آللهای مورد هدف را ارائه میدهد.
۵- تکثیر با PCR و NGS (یا Next-Generation Sequencing) (برای HDR یا NHEJ)
این روش، ارزیابی کمی از ویرایش ژنوم در توالی هدف موردنظر را ارائه میدهد و همچنین میتواند برای بررسی Off-targetها مورد استفاده قرار گیرد.
نتیجهگیری
تکنولوژیهای مبتنی بر CRISPR بدون شک انقلابی را در تحقیقات پایه و همچنین بالینی و بیوتکنولوژی ایجاد کرده است و روز به روز نیز در حال پیشرفت میباشد. در این پست از بلاگ دنازیست آسیا، طور خاص به معرفی تکنولوژی CRISPR و بررسی نکات کاربردی استفاده از آن برای دستکاری ژنوم پرداخته شده است.
منابع
- Adli-2018-The CRISPR tool kit for genome editing and beyond
- CRISPR guide | addgene
- CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular Biology | NEB
- Feature Image | Figure 1 | Figure 2 | Figure 3 | Figure 4 | Figure 5 | Table 1 | Table 2 | Table 3
گردآوری توسط دپارتمان بیولوژی مولکولی شرکت دنازیست آسیا
کیت استخراج پلاسمید (روش ستونی)
۱,۵۱۷,۰۰۰ تومان–۲,۷۰۷,۰۰۰ تومان
{kind=link}
{kind=link}
{kind=link}